
Francis / Goutebroze

Cortical development and pathology
Using genetics, biochemistry and neurobiology, our group has identified certain key causes of severe neurodevelopmental abnormalities. We have questioned the normal functions during development of several cortical proteins (Eml1, Caspr2, Dcx, Dync1h1, Lis1, Rpgrip1l, Dlgap4) associated with the cytoskeleton and the plasma membrane, and effects of their mutation leading to cortical dysplasias.
Summary
Three major proteins mutated in brain malformations or more subtle cortical abnormalities in human have been the focus of our studies in recent years. Cortical malformations are frequent causes of drug-resistant epilepsy and intellectual disability. More subtle abnormalities may be present in idiopathic and mesial temporal lobe epilepsies and in neuropsychiatric disorders. Cortical defects can arise through abnormal neuronal proliferation, migration and/or connectivity. We have used the Eml1, Caspr2 and Dcx proteins as points of entry to better understand normal cortical development and physiopathology. We found that Eml1, likely to be involved in microtubule dynamics, is mutated in severe subcortical heterotopia, associated with mis-positioned neurons in the white matter. Its role in cortical development was previously unknown. Eml1 was studied in neuronal progenitors (e.g. radial glial cells) and immature migrating and differentiating neurons. We identified novel mechanisms leading to heterotopia, including by also identifying Rpgrip1l, Dlgap4 and more recently CerS genes. Mutations in Caspr2, an adhesion protein, are found in a wide spectrum of disorders, including syndromic epilepsy and autism. Better known for its roles at the nodes of Ranvier, its neurodevelopmental functions have been little-studied. It may be essential for adhesive capacities of migrating neurons and during the formation of synapses at the axonal initial segment, hypotheses we are testing. We question the consequences of patient mutations on these functions. DCX, a microtubule-associated protein is mutated in heterotopia and severe brain gyral abnormalities such as lissencephaly. The Dcx knockout is an excellent model to study perturbed subcellular mechanisms, aberrant connectivity and hyperexcitability related to migration defects, epilepsy and behavioral abnormalities. Studying in vivo and human in vitro models, as well as patient mutations (in collaboration with clinicians) for Eml1, Caspr2 and Dcx reveals novel insights into the causes and consequences of abnormal neuronal positioning and connectivity, related to cortical malformations and neuropsychiatric disorders.
Research project
Malformations of cortical development are frequent causes of drug-resistant epilepsy and intellectual disability (1). At least 40% of intractable epilepsy patients are estimated to have severe cortical malformations, and more subtle abnormalities may be underestimated in idiopathic epilepsy, in mesial temporal lobe epilepsy and in neuropsychiatric disorders. Cortical defects can arise through abnormal neuronal proliferation, migration and/or connectivity, and many neurodevelopmental genes play roles in several of these steps. Of interest, some mutated genes can give rise to either severe malformations or more subtle abnormalities in patients, depending on the mutation. The molecular and cellular mechanisms involved in cortex development, and physiopathology related to gene mutations, are still being elucidated. We choose to focus on the roles of three proteins (Eml1, Caspr2, Dcx), which when mutated in patients give rise to a spectrum of cortical dysplasias or neuropsychiatric disorders. We use these proteins as points of entry to better understand key steps of normal and pathological cortical development. Eml1’s role in cortical development is relatively unknown (2-4). We study Eml1 in neuronal progenitors and post-mitotic neurons and question novel mechanisms leading to subcortical heterotopia (Project 1). Defects in the CNTNAP2 gene, coding for the adhesion protein Caspr2, have also recently been characterized in a wide and expanding spectrum of neurodevelopmental disorders, including cortical dysplasia-focal epilepsy syndrome (CDFE) (5) and autism (6). Caspr2 is well-known for its roles in axo-glial contacts at the Nodes of Ranvier (7), but its neurodevelopmental functions are little-studied. We question molecular and cellular functions of Caspr2 during neuronal migration and synaptogenesis, and the consequences of CNTNAP2 variants identified in patients on these functions (Project 2). DCX, a microtubule-associated protein (MAP) which also interacts with membrane proteins, is mutated in heterotopia or severe gyral abnormalities (8,9). We question the subcellular mechanisms, connectivity of aberrantly positioned neurons, origins of hyperexcitability and behavioral deficits in the Dcx knockout (KO) model (Project 3). The aim is to find new strategies for rescuing the phenotypes.
Project 1 : Heterotopia genes and their roles in neuronal progenitors and neurons
Cellular causes of heterotopia have been thought to be primarily due to neuronal migration defects. Eml1 is expressed not only in post-mitotic neurons but also in dividing cells, and ectopic progenitors were identified in Eml1 mutant cortices (Fig 1). Extremely important neuronal progenitors during corticogenesis are radial glial (RG) cells, which give rise to other subtypes of progenitors as well as post-mitotic immature neurons, and serve as guides for migrating neurons (Fig 1, 10). Primary abnormalities in RG cells may hence represent an overlooked cause of heterotopia. We identified mutations in human and mouse Eml1/EML1, showing a role of cortical progenitors in the heterotopia phenotype 2. Mitotic and primary cilia abnormalities were characterized (3; 4; Zaidi et al, submitted). Clinical and bioinformatics studies revealed further heterotopia patients, with mutations in RPGRIP1L and DLGAP4, each gene casting a new light on heterotopia (4; Romero et al., 2022). These are novel data involving stem cells and progenitors which underline new mechanisms for this severe malformation.
General aim: Our strategy is to discover new regulation/dysregulation mechanisms of cortical cell types during corticogenesis (Romero et al, 2018), using disease genes and key models as starting points. Following scientific advances in the field (Klingler et al, 2021), we exploit both mouse and human in vitro models, characterizing these at molecular, subcellular, cellular, anatomical and/or behavioural levels.
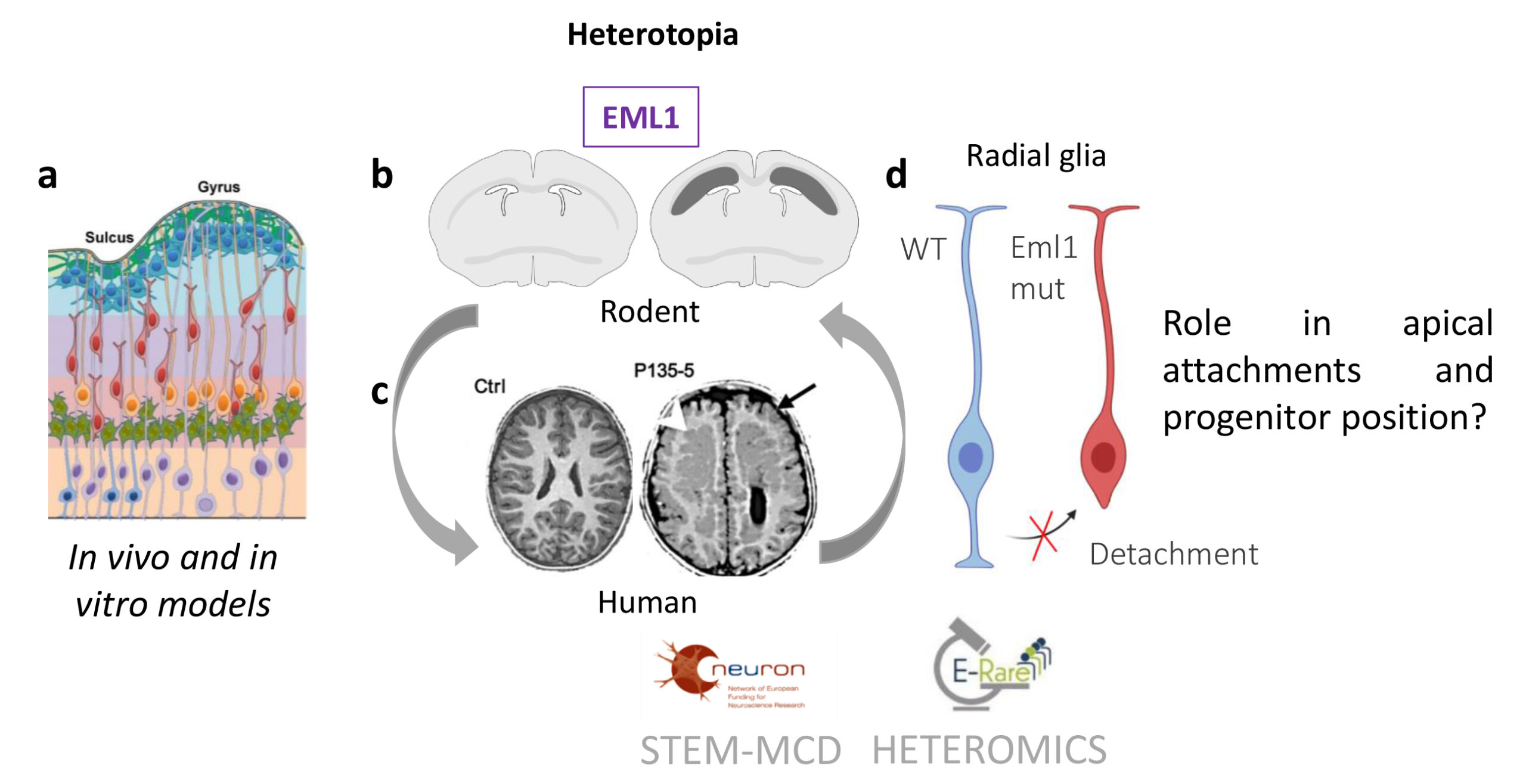
Fig 1. (a) A schema of the developing cortical wall is shown, revealing the different cell types (radial glia, purple; intermediate progenitors, green; basal radial glia, orange; migrating neurons, red; settled neurons in the cortical plate, blue). (b,c) Heterotopia and cell accumulation in the white matter of dorso–medial regions of developing Eml1 mutant (b) and frontal regions of EML1 mutation (c) cortex. (b) The schema shows bilateral bands of subcortical heterotopic neurons (in dark grey on right brain) compared to a wild–type section (left). (c) Control (Ctrl) and EML1 mutation patient (P135-5) MRIs are shown. White arrow head points to the heterotopia. (d) Schema of radial glia cells, in blue a WT cell with apical and basal processes, in red, a mutant cell which has lost its apical attachment.
Project 2 : Characterizing neurodevelopmental roles of Caspr2
Heterozygous copy-number variations, missense variants and homozygous defects in CNTNAP2 have repeatedly been reported as susceptibility factors in developmental language delay, autism spectrum disorders, Gilles de la Tourette syndrome, epilepsy, CDFE, schizophrenia, and severe intellectual disability5,6,12-15. Analyses of KO mice (Cntnap2-/-) revealed impaired migration of cortical projection neurons and a reduced number of GABAergic interneurons, associated with asynchronous cortical activity and epileptic seizures16. Thus Caspr2 plays a critical role in neuronal migration. Furthermore, Caspr2 is enriched at the axon initial segments (AIS) of pyramidal cells from human temporal cortex (Fig 2), which act as sites for action potential initiation and are master integrators of synaptic events regulating excitability17,18. In the mammalian cerebral cortex, AIS are contacted by the axons of Chandelier type parvalbumin-positive GABAergic interneurons (Ch). Thus Caspr2 may be involved in the development and/or function of synapses between Ch and pyramidal cell AIS, and the mutations identified in patients may lead to hyperexcitability and network disfunction.
General aim : To elucidate essential roles of Caspr2 in immature neurons taking advantage of the availability of Cntnap2 mutant mice.
Specific aims : We plan: (a) to characterize fundamental molecular and cellular functions of Caspr2 during neuronal migration and synaptogenesis using in vivo (in utero electroporation) and in vitro (primary neurons and brain slices cultures; immunochemistry and time-lapse videomicroscopy, biochemistry) approaches; and (b) to determine the pathological consequences of mutations identified in patients on these functions.
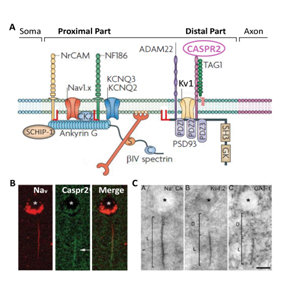
Fig. 2. (A) AIS molecular complexes composed of ion channels (Nav, KCNQ2-3, Kv1), cell adhesion molecules and cytoskeletal scaffolds (adapted from17) (B). Immunolocalization of Nav and Caspr2 (arrows) at the AIS of a pyramidal cell (PC) from the human temporal cortex. (C) Photomicrographs showing the AIS of a PC immuno stained for Nav (Na+ Ch), Kv1.2 subunits and the GABA transporter GAT-1. (B, C, adapted from19).
Project 3 : Characterizing the origins, features and consequences of hyperexcitability in Dcx knockout (KO) cells (Richard Belvindrah)
All type I lissencephaly gene mouse models show hippocampal heterotopia and more widespread interneuron defects as common features (20-24 and unpublished). In Dcx KO mice, CA3 hippocampal pyramidal cells are abnormally positioned (Fig 3) and this is associated with altered dendritic form, mossy fiber pruning defects, hyperexcitability and spontaneous epilepsy 25,26. We hypothesize that aberrant position, connectivity and intrinsic defects of Dcx KO cells contribute to hyperexcitability. Although it is known that Dcx is a microtubule-associated protein (MAP, e.g. 9, 27), the molecular and cellular mechanisms leading to this phenotype and more generally behavioural and neuropsychiatric deficits, are still not clear in Dcx KO mice which represent a well-characterized tool to further study this.
Omics and anatomical data reveal an inversion of hippocampal pyramidal layers (CA3 region) and unexpected organelle abnormalities in Dcx mice (28-30). Dcx is developmentally-regulated and not expressed in mature neurons, however organelle abnormalities persist in the adult (30). This is a first report of such organelle defects in pyramidal neurons, likely to impact function, contributing to neurodevelopmental neuronal migration disorders.
General aims: We plan to study (a) the link between abnormal organelles (e.g. mitochondria), disrupted migration, neuronal position, connectivity and function; (b) intrinsic factors in adult hippocampal neurons affecting abnormal function; and (c) strategies during development for improving migration and reducing hyperexcitability of abnormal Dcx KO cells.
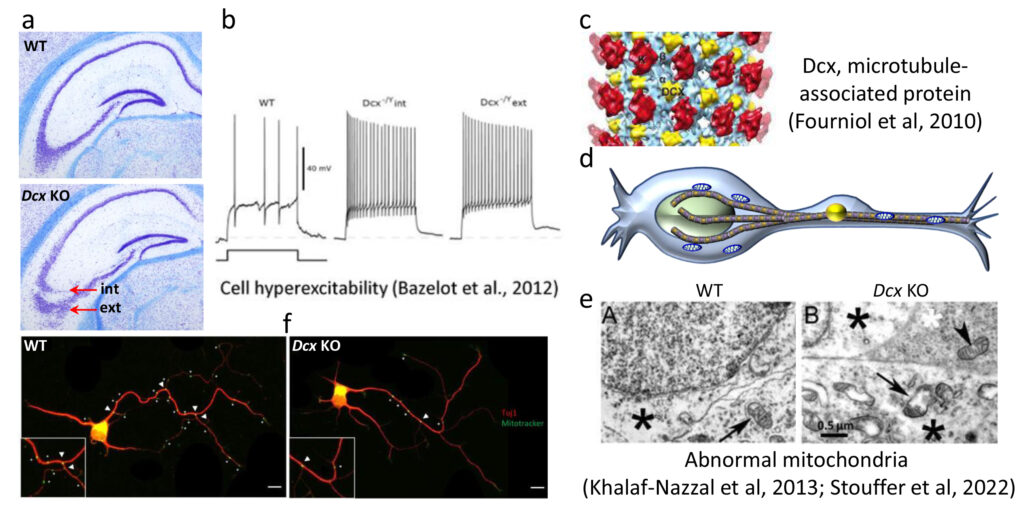
Fig 3. Hippocampal heterotopia in Dcx KO mice and role of Dcx influencing mitochondria. (a) Two CA3 pyramidal cell layers (internal and external) are observed (red arrows). (b) Hyperexcitability is associated with both layers 26, (c) Dcx (yellow) is a microtubule associated protein (27), (d) schematic showing mitochondria (blue) on microtubules (brown) in a migrating neuron; (e) abnormal mitochondria are observed by electron microscopy in Dcx KO neurons (28); (f) Mitotracker labelling in hippocampal neurons in culture shows fewer mitochondria at branchpoints (30).
References
1 Guerrini, R., Dobyns, W.B., & Barkovich, A.J. (2008) Trends Neurosci 31, 154-162.
2 Kielar et al. Nat Neurosci 2014 17, 923-933.
3 Bizzotto et al. Sci Rep. 2017 Dec 11;7(1):17308.
4 Uzquiano et al. Cell Rep. 2019;28:1596-1611.
5 Strauss, K.A., Puffenberger, E.G., Huentelman, M.J. et al. (2006) N Engl J Med 354, 1370-1377.
6 Penagarikano, O. & Geschwind, D.H. (2012) Trends Mol Med 18, 156-163.
7 Susuki, K. & Rasband, M.N. (2008) Curr Opin Cell Biol 20, 616-623.
8 des Portes, V., Pinard, J.M., Billuart, P. et al. (1998) Cell 92, 51-61.
9 Francis, F., Koulakoff, A., Boucher, D. et al. (1999) Neuron 23, 247-256.
10 Noctor, S.C., Martinez-Cerdeno, V., & Kriegstein, A.R. (2007) Novartis Found Symp 288, 59-73; discussion 73-58, 96-58.
11 Hansen, D.V., Lui, J.H. Parker, P.R.L., & Kriegstein, A.R. (2010) Nature 464, 554-561.
12 Verkerk, A.J., Mathews, C.A., Joosse, M. et al. (2003) Genomics 82, 1-9.
13 Friedman, J.I., Vrijenhoek, T., Markx, S. et al. (2008) Mol Psychiatry 13, 261-266.
14 Zweier, C., de Jong, E.K., Zweier, M. et al. (2009) Am J Hum Genet 85, 655-666.
15 Gregor, A., Albrecht, B., Bader, I. et al. (2011) BMC Med Genet 12, 106.
16 Penagarikano, O., Abrahams, B.S., Herman, E.I. et al. (2011) Cell 147, 235-246.
17 Rasband, M.N. (2010) Nat Rev Neurosci 11, 552-562.
18 Kole, M.H. & Stuart, G.J. (2012) Neuron 73, 235-247.
19 Inda, M.C., DeFelipe, J., & Munoz, A. (2006) Proc Natl Acad Sci U S A 103, 2920-2925.
20 Corbo, J.C., Deuel, T.A., Long, J.M. et al. (2002) J Neurosci 22, 7548-7557.
21 Kappeler, C., Saillour, Y., Baudoin, J.P. et al. (2006) Hum Mol Genet 15, 1387-1400.
22 Fleck, M.W., Hirotsune, S., Gambello, M.J. et al. (2000) J Neurosci 20, 2439-2450.
23 Nasrallah, I.M., McManus, M.F., Pancoast, M.M. et al. (2006) J Comp Neurol 496, 847-858.
24 Keays, D.A., Tian, G., Poirier, K. et al. (2007) Cell 128, 45-57.
25 Nosten-Bertrand, M., Kappeler, C., Dinocourt, C. et al. (2008) PLoS One 3, e2473.
26 Bazelot, M., Simonnet, J., Dinocourt, C. et al. (2012) Eur J Neurosci 35, 244-256.
27 Fourniol, F et al (2010) J Cell Biol. Nov 1;191(3):463-70.
28 Khalaf-Nazzal, R et al (2013) PLoS One. Sep 2;8(9):e72622.
29 Khalaf-Nazzal, R, Stouffer, M.A. et al (2017) Hum Mol Genet. Jan 1;26(1):90-108.
30 Stouffer, M.A., Khalaf-Nazzal, R., (2022). Neurobiol Dis 168, 105702.
Team composition
- Group leader: Fiona Francis, DRCE CNRS (PhD, HDR)
- Laurence Goutebroze, DR2 CNRS (PhD, HDR)
- Marika Nosten-Bertrand, CRCN CNRS (PhD)
- Marta Garcia, MC SU (PhD)
- Richard Belvindrah, MC HDR SU (PhD)
- Gael Grannec, AI SU
- Taylor Manett, PhD student
- Kaviya Chinnappa, Post-doc
- Loïc Angrand, PhD student (ENVA)
- Valeria Viola, PhD student
- Alexandra Chrétien, PhD student (ENVA)
- Fiona Ballorin, PhD student
- Cloé Bourel, IE CDD
Collaborations
- N. Bahi-Buisson Institut Imagine Paris France
- J. Ladewig HITBR Mannheim Germany
- S. Cappello LMU Munich Germany
- D. Jabaudon Univ Geneva Switzerland
- V. Borrell Institute Neuroscience Alicante Spain
- J-L Mandel IGBMC, Strasbourg France
- J-B Manent INMED Marseille France
- N. Ozlu Koc University Turkey
- M. Cohen-Salmon CIRB Paris France
- C. Plisson-Chastang CBI Toulouse France
- S. Lebaron CBI Toulouse France
- A. Baffet Institut Curie Paris France
- J. Livet Institut de la Vision Paris France
- C. Moores Birkbeck College London UK
- S Bonneau, LJP, IBPS, France
- C. Depienne ICM Paris France
- C. Faivre-Sarrailh CRN2M Marseille France
- B. Dargent CRN2M Marseille France
Funding
Equipe FRM, ANR, Era-net E-RARE, FRC, SU-Emergence, Prix Valérie Chamaillard, ARSEP, Brixham Foundation
Previous: ANR, Era-net, FP7, Fondation Bettencourt Schueller, Fondation Orange, Labex Bio-Psy
A class-specific effect of dysmyelination on the excitability of hippocampal interneurons
Pinatel D, Pearlstein E, Bonetto G, Goutebroze L, Karagogeos D, Crepel V, Faivre-Sarrailh C.
Elife. 2023 Oct 16:12:e86469.
PMID:37843188 ![]()
Grey matter heterotopia subtypes show specific morpho-electric signatures and network dynamics
Vermoyal JC, Hardy D, Goirand-Lopez L, Vinck A, Silvagnoli L, Fortoul A, Francis F, Cappello S, Bureau I, Represa A, Cardoso C, Watrin F, Marissal T, Manent JB.
Brain. 2023 Sep 19:awad318.
PMID:37724593 ![]()
A human dynein heavy chain mutation impacts cortical progenitor cells causing developmental defects, reduced brain size and altered brain architecture
Romero DM, Zaidi D, Cifuentes-Diaz C, Maillard C, Grannec G, Selloum M, Birling MC, Bahi-Buisson N, Francis F
Neurobiol Dis. 2023 May;180:106085.
PMID:36933672 ![]()
Differential impacts of Cntnap2 heterozygosity and Cntnap2 null homozygosity on axon and myelinated fiber development in mouse
Cifuentes-Diaz C, Canali G, Garcia M, Druart M, Manett T, Savariradjane M, Guillaume C, Le Magueresse C, Goutebroze L
Front Neurosci. 2023 Jan 30;17:1100121.
PMID:36793543 ![]()
Primary Cilia Influence Progenitor Function during Cortical Development.
Zaidi D, Chinnappa K, Francis F.
Cells. 2022 Sep 16;11(18):2895.
PMID:36139475 ![]()
Inflammation and Autophagy: A Convergent Point between Autism Spectrum Disorder (ASD)-Related Genetic and Environmental Factors: Focus on Aluminum Adjuvants.
Angrand L, Masson JD, Rubio-Casillas A, Nosten-Bertrand M, Crépeaux G.
Toxics. 2022 Aug 31;10(9):518.
PMID:36136483 ![]()
RAB6 and dynein drive post-Golgi apical transport to prevent neuronal progenitor delamination
Brault JB, Bardin S, Lampic M, Carpentieri JA, Coquand L, Penisson M, Lachuer H, Victoria GS, Baloul S, El Marjou F, Boncompain G, Miserey-Lenkei S, Belvindrah R, Fraisier V, Francis F, Perez F, Goud B, Baffet AD
EMBO Rep. 2022 Aug 18:e54605
PMID:35979738 ![]()
Novel role of the synaptic scaffold protein Dlgap4 in ventricular surface integrity and neuronal migration during cortical development
Romero DM, Poirier K, Belvindrah R, Moutkine I, Houllier A, LeMoing AG, Petit F, Boland A, Collins SC, Soiza-Reilly M, Yalcin B, Chelly J, Deleuze JF, Bahi-Buisson N, Francis F
Nat Commun. (2022) 13, 2746
PMID:35585091 ![]()
Visualising the cytoskeletal machinery in neuronal growth cones using cryo-electron tomography
Atherton J, Stouffer M, Francis F, Moores CA
J Cell Sci. (2022) 135
PMID:35383828 ![]()
Doublecortin mutation leads to persistent defects in the Golgi apparatus and mitochondria in adult hippocampal pyramidal cells
Stouffer MA, Khalaf-Nazzal R, Cifuentes-Diaz C, Albertini G, Bandet E, Grannec G, Lavilla V, Deleuze JF, Olaso R, Nosten-Bertrand M, Francis F
Neurobiol Dis. (2022) 168, 105702
PMID:35339680 ![]()