
Francis / Goutebroze

Développement cortical et pathologie
Grâce à la génétique, la biochimie et la neurobiologie, notre groupe a identifié certaines causes clés d’anomalies neurodéveloppementales sévères. Nous nous sommes interrogés sur les fonctions normales au cours du développement de plusieurs protéines corticales (Eml1, Caspr2, Dcx, Dync1h1, Lis1, Rpgrip1l, Dlgap4) associées au cytosquelette et à la membrane plasmique, et sur les effets de leur mutation conduisant à des dysplasies corticales.
Résumé
Trois protéines majeures mutées dans des malformations cérébrales, ou des anomalies corticales plus subtiles, chez l’homme ont fait l’objet de nos études ces dernières années. Les malformations corticales constituent une cause fréquente d’épilepsie résistante aux traitements et de déficience intellectuelle. Des anomalies plus subtiles ont potentiellement un impact sur la manifestation d’épilepsies idiopathiques, d’épilepsies des lobes temporaux moyens et de troubles neuropsychiatriques. Les défauts corticaux peuvent résulter d’anomalies de prolifération, de migration et/ou de connectivité neuronale. Nous avons utilisé les trois protéines Eml1, Caspr2 et Dcx, comme points d’entrée pour mieux comprendre le développement cortical normal et sa physiopathologie. Nous avons identifié des mutations de la protéine Eml1, impliquée dans la dynamique des microtubules, dans des cas d’hétérotopies sous-corticales sévères, avec une localisation anormale de neurones dans la substance blanche. Son rôle dans le développement cortical était jusqu’à présent inconnu. Eml1 a été étudié dans les progéniteurs neuronaux de la souris (par exemple, les cellules gliales radiales) et les neurones immatures en migration et en différenciation. Nous avons identifié de nouveaux mécanismes conduisant à l’hétérotopie, notamment en identifiant également les gènes Rpgrip1l, Dlgap4 et plus récemment CerS. Des mutations de Caspr2, une protéine d’adhérence, ont été identifiées dans un large spectre de maladies incluant l’épilepsie et l’autisme. Le rôle de la protéine Caspr2 dans l’organisation des nœuds de Ranvier est bien caractérisé mais ses fonctions neurodéveloppementales ont été peu étudiées. Cette dernière pourrait jouer un rôle essentiel dans les propriétés adhésives des neurones en migration et au cours de la formation des synapses au niveau du segment initial, hypothèses que nous testons. Nous étudions les conséquences des mutations identifiées chez les patients sur ces fonctions. DCX, une protéine associée aux microtubules, est mutée dans des cas d’hétérotopies et d’anomalies sévères de gyration du cerveau telles que les lissencéphalies. Le knock-out Dcx est un excellent modèle pour étudier les mécanismes subcellulaires perturbés, la connectivité aberrante et l’hyperexcitabilité liées aux défauts de migration, à l’épilepsie et aux anomalies comportementales. L’étude de modèles in vivo et in vitro humains, ainsi que de mutations de patients (en collaboration avec des cliniciens) pour Eml1, Caspr2 et Dcx, permet de mieux comprendre les causes et les conséquences d’un positionnement et d’une connectivité neuronaux anormaux, liés à des malformations corticales et à des troubles neuropsychiatriques.
Projets de recherche
Les malformations corticales sont une cause fréquente d’épilepsies résistantes aux traitements et de déficits intellectuels (1). Au moins 40% des patients souffrant d’épilepsie intraitable présentent de sévères malformations corticales, et le nombre d’anomalies plus subtiles est probablement sous-estimé dans les cas d’épilepsies idiopathiques, d’épilepsies des lobes temporaux moyens et les maladies neuropsychiatriques (e.g. et voir ci-dessous). Les défauts corticaux peuvent résulter d’une prolifération, migration et/ou connectivité neuronale anormale, et de nombreux gènes neurodéveloppementaux jouent un rôle au niveau de ces différentes étapes. De façon intéressante, certains gènes mutés sont associés soient à des malformations sévères soient à des anomalies plus subtiles chez les patients, en fonction de la mutation. Les mécanismes moléculaires et cellulaires impliqués dans le développement cortical, et la physiopathologie associée aux mutations, restent à être élucider. Nous avons choisi de nous concentrer sur le rôle de trois protéines (Eml1, Caspr2 et Dcx) qui, lorsqu’ils sont mutés chez les patients, donnent lieu à un éventail de dysplasies corticales ou de troubles neuropsychiatriques. Nous utilisons ces protéines comme points d’entrée pour mieux comprendre les étapes clés du développement cortical normal et pathologique. Le rôle d’Eml1 dans le développement cortical est actuellement inconnu (2-4). Nous étudions son rôle dans les progéniteurs neuronaux et les neurones, et les mécanismes conduisant à l’hétérotopie sub-corticale (Projet 1). Des mutations du gène CNTNAP2 codant pour la protéine d’adhésion Caspr2, ont également été identifiées récemment dans un spectre large et croissant de maladies neurodéveloppementales incluant le syndrome d’épilepsie « cortical dysplasia-focal epilepsy » (CDFE) (5) et l’autisme (6). Le rôle de Caspr2 dans la formation des contacts axogliaux au niveau des noeuds de Ranvier est bien connu, mais ses fonctions au cours du développement ont été peu étudiées. Nous évaluons les fonctions de Caspr2 au cours de la migration neuronale et de la synaptogénèse, et les conséquences des variants de CNTNAP2 identifiés chez les patients sur ces fonctions (Projet 2). DCX, une protéine associée aux microtubules (MAP), est mutée dans des cas d’hétérotopies sub-corticales et des anomalies sévères de gyration (8,9). Nous étudions les neurones positionnés de façon aberrante, l’origine de l’hyperexcitabilité et les anomalies de comportement des modèles pour le gène Dcx (KO) (Projet 3). L’objectif est de trouver de nouvelles stratégies pour sauver les phénotypes.
Projet 1 : Les gènes d’hétérotopies et leurs rôles dans les progéniteurs neuronaux et les neurones
Les résultats de la littérature suggéraient jusqu’à récemment que l’hétérotopie était principalement la conséquence directe de défauts de migration des neurones corticaux. Eml1 est exprimée dans les neurones post-mitotiques et dans les cellules en division. Des progéniteurs présentant une localisation ectopique ont été identifiés au niveau du cortex dans un modèle présentant une mutation du gène Eml1 (Fig 1). Les cellules gliales radiaires (RG) sont des neurones progéniteurs extrêmement importants lors de la corticogenèse, qui donnent naissance à d’autres sous-types de progéniteurs, et de neurones immatures post-mitotiques, et servent de guides pour les neurones en migration (Fig 1, 10). Les anomalies primaires des cellules RG peuvent donc représenter une cause négligée d’hétérotopie. Nous avons identifié des mutations dans Eml1/EML1 chez l’homme et la souris, montrant un rôle des progéniteurs corticaux dans le phénotype de l’hétérotopie (2). Des anomalies mitotiques et des cils primaires ont été caractérisés (3 ;4 ; Zaidi et al, submitted). Des études cliniques et bioinformatiques ont révélé d’autres patients atteints d’hétérotopie, avec des mutations dans RPGRIP1L et DLGAP4, chaque gène jetant un nouvel éclairage sur l’hétérotopie (3; Romero et al., 2022). Il s’agit de nouvelles données impliquant les cellules souches et les progéniteurs qui soulignent de nouveaux mécanismes pour cette malformation sévère.
Notre stratégie consiste à découvrir de nouveaux mécanismes de régulation/dérégulation des types de cellules corticales au cours de la corticogenèse (Romero et al, 2018), en utilisant des gènes de maladies et des modèles clés comme points de départ. Suite aux avancées scientifiques dans le domaine (Klingler et al, 2021), nous exploitons des modèles in vitro à la fois murins et humains, en les caractérisant aux niveaux moléculaire, subcellulaire, cellulaire, anatomique et/ou comportementale.
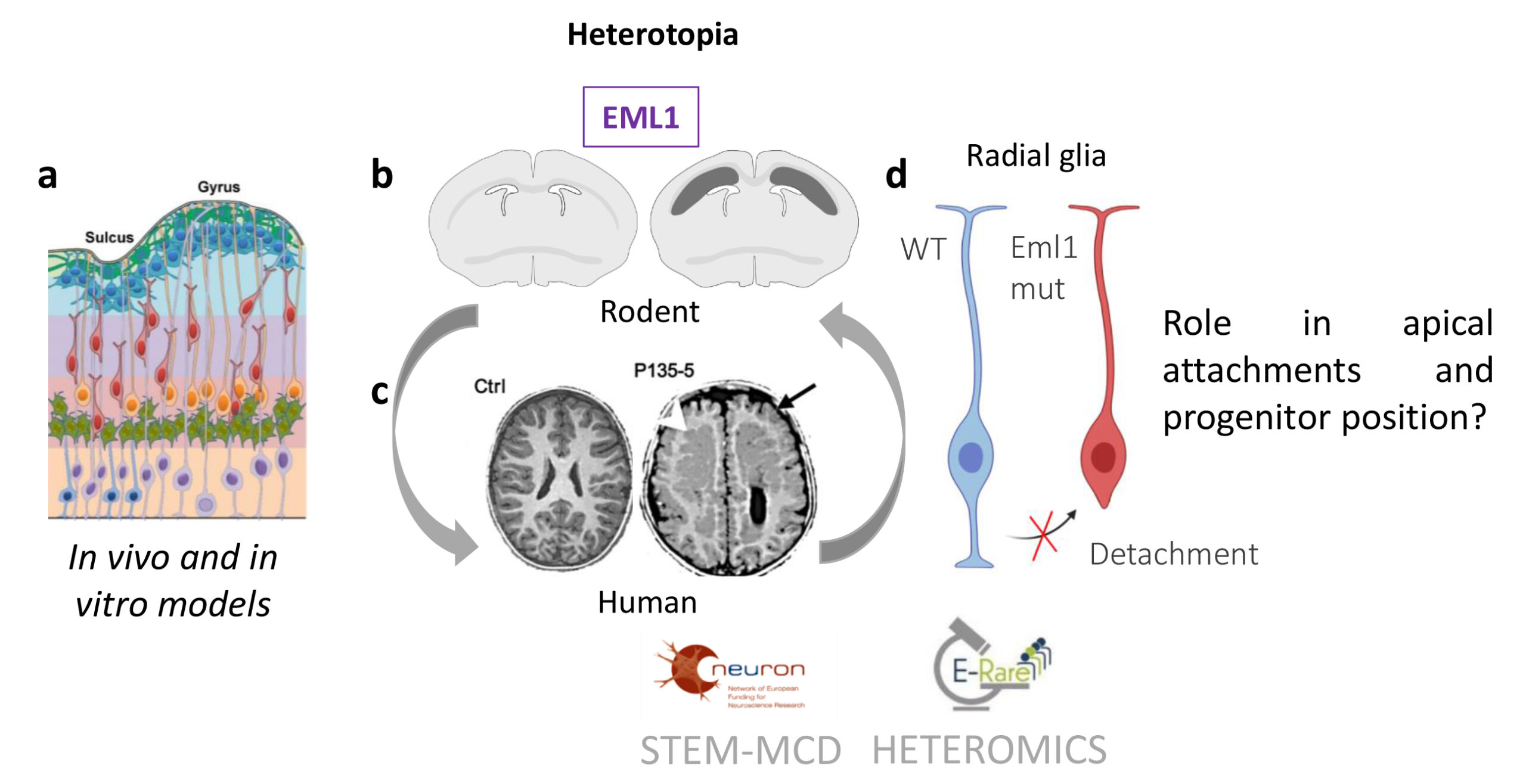
Fig 1. (a) Un schéma de la paroi corticale en développement est montré, révélant les différents types de cellules (glie radiale, violet ; progéniteurs intermédiaires, vert ; glie radiale basale, orange ; neurones en migration, rouge ; neurones installés dans la plaque corticale, bleu). (b,c) Hétérotopie et accumulation de cellules dans la substance blanche des régions dorso-médianes du cortex en développement du mutant Eml1 (b) et des régions frontales du mutant EML1 (c). (b) Le schéma montre des bandes bilatérales de neurones hétérotopiques sous-corticaux (en gris foncé sur le cerveau droit) par rapport à une section de type sauvage (à gauche). (c) Des IRM de patients témoins (Ctrl) et de patients atteints de la mutation EML1 (P135-5) sont montrées. La flèche blanche pointe vers l’hétérotopie. (d) Schéma des cellules de la glie radiale, en bleu une cellule WT avec des processus apicaux et basaux, en rouge une cellule mutante qui a perdu son attachement apical.
Projet 2 : Caractérisation des rôles développementaux de Caspr2
De nombreuses anomalies génétiques (chromosomiques et mutations faux-sens hétérozygotes; altérations homozygotes) du gène CNTNAP2 ont été reportées comme étant des facteurs de susceptibilité chez des patients atteints de retard de langage, d’autisme, de syndrome de Gilles de la Tourette, d’épilepsie, de CDFE, de schizophrénie et de déficience intellectuelle sévère5,6,12-15. Les analyses des souris invalidées pour l’expression de Cntnap2 (Cntnap2-/-) ont révélé des altérations de migration des neurones de projection corticaux et un nombre réduit d’interneurones GABAergiques, associés à une activité corticale asynchrone et des crises d’épilepsie16. Caspr2 semble donc jouer un rôle critique dans la migration neuronale. De plus Caspr2 est enrichie au niveau des segments initiaux axonaux (SIA) des cellules pyramidales du cortex temporal humain (Fig 2) qui jouent le rôle de sites d’initiation des potentiels d’action et sont des intégrateurs majeurs des évènements synaptiques régulant l’excitabilité17,18. Dans le cortex cérébral des mammifères, les SIA sont en contact avec les axones des interneurones GABAergiques de type Chandelier exprimant la parvalbumine. De fait, Caspr2 pourrait également intervenir dans la mise en place et/ou la fonction des synapses entre les SIA des cellules pyramidales et les cellules Chandelier, et les mutations identifiées chez les patients pourraient conduire à une hyperexcitabilité et un dysfonctionnement du réseau neuronal.
Notre objectif principal est d’investiguer les rôles essentiels de Caspr2 dans les neurones immatures, en profitant de la disponibilité des souris Cntnap2-/- au laboratoire.
Nos objectifs spécifiques sont de: (a) Caractériser les fonctions cellulaires et moléculaires fondamentales de Capsr2 au cours de la migration neuronale et de la synaptogénèse par des approches combinées in vivo (électroporation in utero) et in vitro (cultures de neurones primaires et de tranches de cerveaux ; immunohistochimie et vidéomicroscopie ; biochimie) ; (b) Déterminer les conséquences pathologiques des mutations identifiées chez les patients sur ces différentes fonctions.
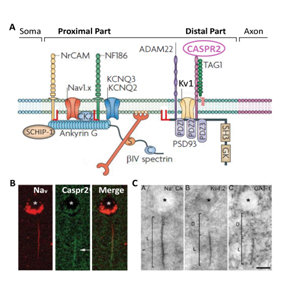
Fig 2. (A) Complexes moléculaires du SIA composés de canaux ioniques (Nav, KCNQ2-3, Kv1), de molécules d’adhésion cellulaire et de protéines d’échaffaudage du cytosquelette (adapté à partir de 17) (B). Immunolocalisation des canaux Nav et de Caspr2 (flèches) au niveau du SIA d’une cellule pyramidale du cortex temporal humain. (C) Photomicrographies montrant les SIA de cellules pyramidales marqués pour les canaux Nav (Na+ Ch), les sous-unités Kv1.2 et le transporteur du GABA GAT-1. (B, C, adapté à partir de 19).
Projet 3 : Identification des origines, caractéristiques et conséquences de l’hyperexcitabilité des cellules invalidées pour l’expression du gène Dcx (Dcx KO) (Richard Belvindrah)
Tous les modèles murins de lissencephalie de type I présentent comme caractéristiques communes une hétérotopie hippocampique et des défauts d’interneurones plus généralisée (20-24 et observations non publiées). Chez les souris Dcx KO, les cellules pyramidales de la région CA3 de l’hippocampe présentent une localisation anormale (Fig 3) associée à une forme dendritique altérée, des anomalies subtiles de connectivité des fibres moussues, une hyperexcitabilité et une épilepsie spontanée (25,26). Notre hypothèse est que la position anormale, la connectivité aberrante et les anomalies intrinsèques des cellules Dcx KO contribuent à l’hyperexcitabilité. Bien que l’on sache que Dcx est une protéine associée aux microtubules (MAP, par exemple 9, 27), les mécanismes exacts conduisant à ce phénotype, et plus généralement aux défauts de comportement et neuropsychiatriques des souris Dcx KO, ne sont pas encore clairs.
Les données omiques et anatomiques révèlent une inversion des couches pyramidales de l’hippocampe (région CA3) et des anomalies inattendues des organites chez les souris Dcx (28-30). Dcx est régulée pendant le développement et n’est pas exprimée dans les neurones matures, mais les anomalies des organites persistent chez l’adulte (30). Il s’agit d’une première description de telles anomalies des organites dans les neurones pyramidaux, susceptibles d’avoir un impact sur la fonction, en contribuant aux troubles de la migration neuronale au cours du développement neurologique.
Nos objectifs principaux sont d’étudier: (a) le lien entre les organites anormaux (par exemple les mitochondries), la migration, la position, la connectivité et la fonction perturbée des neurones ; (b) les facteurs intrinsèques dans les neurones de l’hippocampe adulte affectant leur fonction; (c) des stratégies pendant le développement permettant d’améliorer la migration et visant à réduire l’hyperexcitabilité cellulaire.
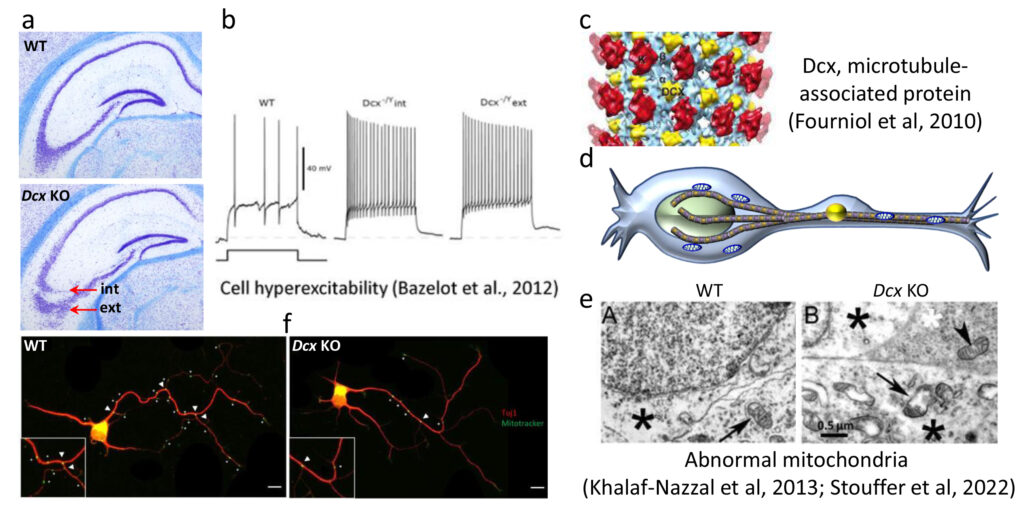
Fig 3. Hétérotopie hippocampique chez les souris Dcx KO et rôle de Dcx influençant les mitochondries. (a) Deux couches (interne et externe) de cellules pyramidales CA3 sont observées (flèches rouges); (b) l’hyperexcitabilité est associée aux deux couches (26); (c) Dcx (jaune) est une protéine associée aux microtubules (27); (d) schéma montrant les mitochondries (bleu) sur les microtubules (marron) dans un neurone en migration; (e) des mitochondries anormales sont observées par microscopie électronique dans les neurones Dcx KO (28); (f) le marquage Mitotracker dans les neurones hippocampiques en culture montre une diminution des mitochondries aux points d’embranchements (30).
References
1 Guerrini, R., Dobyns, W.B., & Barkovich, A.J. (2008) Trends Neurosci 31, 154-162.
2 Kielar et al. Nat Neurosci 2014 17, 923-933.
3 Bizzotto et al. Sci Rep. 2017 Dec 11;7(1):17308.
4 Uzquiano et al. Cell Rep. 2019;28:1596-1611.
5 Strauss, K.A., Puffenberger, E.G., Huentelman, M.J. et al. (2006) N Engl J Med 354, 1370-1377.
6 Penagarikano, O. & Geschwind, D.H. (2012) Trends Mol Med 18, 156-163.
7 Susuki, K. & Rasband, M.N. (2008) Curr Opin Cell Biol 20, 616-623.
8 des Portes, V., Pinard, J.M., Billuart, P. et al. (1998) Cell 92, 51-61.
9 Francis, F., Koulakoff, A., Boucher, D. et al. (1999) Neuron 23, 247-256.
10 Noctor, S.C., Martinez-Cerdeno, V., & Kriegstein, A.R. (2007) Novartis Found Symp 288, 59-73; discussion 73-58, 96-58.
11 Hansen, D.V., Lui, J.H. Parker, P.R.L., & Kriegstein, A.R. (2010) Nature 464, 554-561.
12 Verkerk, A.J., Mathews, C.A., Joosse, M. et al. (2003) Genomics 82, 1-9.
13 Friedman, J.I., Vrijenhoek, T., Markx, S. et al. (2008) Mol Psychiatry 13, 261-266.
14 Zweier, C., de Jong, E.K., Zweier, M. et al. (2009) Am J Hum Genet 85, 655-666.
15 Gregor, A., Albrecht, B., Bader, I. et al. (2011) BMC Med Genet 12, 106.
16 Penagarikano, O., Abrahams, B.S., Herman, E.I. et al. (2011) Cell 147, 235-246.
17 Rasband, M.N. (2010) Nat Rev Neurosci 11, 552-562.
18 Kole, M.H. & Stuart, G.J. (2012) Neuron 73, 235-247.
19 Inda, M.C., DeFelipe, J., & Munoz, A. (2006) Proc Natl Acad Sci U S A 103, 2920-2925.
20 Corbo, J.C., Deuel, T.A., Long, J.M. et al. (2002) J Neurosci 22, 7548-7557.
21 Kappeler, C., Saillour, Y., Baudoin, J.P. et al. (2006) Hum Mol Genet 15, 1387-1400.
22 Fleck, M.W., Hirotsune, S., Gambello, M.J. et al. (2000) J Neurosci 20, 2439-2450.
23 Nasrallah, I.M., McManus, M.F., Pancoast, M.M. et al. (2006) J Comp Neurol 496, 847-858.
24 Keays, D.A., Tian, G., Poirier, K. et al. (2007) Cell 128, 45-57.
25 Nosten-Bertrand, M., Kappeler, C., Dinocourt, C. et al. (2008) PLoS One 3, e2473.
26 Bazelot, M., Simonnet, J., Dinocourt, C. et al. (2012) Eur J Neurosci 35, 244-256.
27 Fourniol, F et al (2010) J Cell Biol. Nov 1;191(3):463-70.
28 Khalaf-Nazzal, R et al (2013) PLoS One. Sep 2;8(9):e72622.
29 Khalaf-Nazzal, R, Stouffer, M.A. et al (2017) Hum Mol Genet. Jan 1;26(1):90-108.
30 Stouffer, M.A., Khalaf-Nazzal, R., (2022). Neurobiol Dis 168, 105702.
Composition de l’équipe
- Responsable d’équipe : Fiona Francis, DRCE CNRS (PhD, HDR)
- Laurence Goutebroze, DR2 CNRS (PhD, HDR)
- Marika Nosten-Bertrand, CRCN CNRS (PhD)
- Marta Garcia, MC SU (PhD)
- Richard Belvindrah, MC HDR SU (PhD)
- Gael Grannec, AI SU
- Taylor Manett, étudiante en thèse
- Kaviya Chinnappa, Post-doc
- Loïc Angrand, étudiant en thèse (ENVA)
- Valeria Viola, étudiante en thèse
- Alexandra Chrétien, étudiante en thèse (ENVA)
- Fiona Ballorin, étudiante en thèse
- Cloé Bourel, IE CDD
Collaborations
- N. Bahi-Buisson Institut Imagine Paris France
- J. Ladewig HITBR Mannheim Germany
- S. Cappello LMU Munich Germany
- D. Jabaudon Univ Geneva Switzerland
- V. Borrell Institute Neuroscience Alicante Spain
- J-L Mandel IGBMC, Strasbourg France
- J-B Manent INMED Marseille France
- N. Ozlu Koc University Turkey
- M. Cohen-Salmon CIRB Paris France
- C. Plisson-Chastang CBI Toulouse France
- S. Lebaron CBI Toulouse France
- A. Baffet Institut Curie Paris France
- J. Livet Institut de la Vision Paris France
- C. Moores Birkbeck College London UK
- S Bonneau, LJP, IBPS, France
- C. Depienne ICM Paris France
- C. Faivre-Sarrailh CRN2M Marseille France
- B. Dargent CRN2M Marseille France
Financements
Equipe FRM, ANR, Era-net E-RARE, FRC, SU-Emergence, Prix Valérie Chamaillard, ARSEP, Brixham Foundation
Previous: ANR, Era-net, FP7, Fondation Bettencourt Schueller, Fondation Orange, Labex Bio-Psy
A class-specific effect of dysmyelination on the excitability of hippocampal interneurons
Pinatel D, Pearlstein E, Bonetto G, Goutebroze L, Karagogeos D, Crepel V, Faivre-Sarrailh C.
Elife. 2023 Oct 16:12:e86469.
PMID:37843188 ![]()
Grey matter heterotopia subtypes show specific morpho-electric signatures and network dynamics
Vermoyal JC, Hardy D, Goirand-Lopez L, Vinck A, Silvagnoli L, Fortoul A, Francis F, Cappello S, Bureau I, Represa A, Cardoso C, Watrin F, Marissal T, Manent JB.
Brain. 2023 Sep 19:awad318.
PMID:37724593 ![]()
A human dynein heavy chain mutation impacts cortical progenitor cells causing developmental defects, reduced brain size and altered brain architecture
Romero DM, Zaidi D, Cifuentes-Diaz C, Maillard C, Grannec G, Selloum M, Birling MC, Bahi-Buisson N, Francis F
Neurobiol Dis. 2023 May;180:106085.
PMID:36933672 ![]()
Differential impacts of Cntnap2 heterozygosity and Cntnap2 null homozygosity on axon and myelinated fiber development in mouse
Cifuentes-Diaz C, Canali G, Garcia M, Druart M, Manett T, Savariradjane M, Guillaume C, Le Magueresse C, Goutebroze L
Front Neurosci. 2023 Jan 30;17:1100121.
PMID:36793543 ![]()
Primary Cilia Influence Progenitor Function during Cortical Development.
Zaidi D, Chinnappa K, Francis F.
Cells. 2022 Sep 16;11(18):2895.
PMID:36139475 ![]()
Inflammation and Autophagy: A Convergent Point between Autism Spectrum Disorder (ASD)-Related Genetic and Environmental Factors: Focus on Aluminum Adjuvants.
Angrand L, Masson JD, Rubio-Casillas A, Nosten-Bertrand M, Crépeaux G.
Toxics. 2022 Aug 31;10(9):518.
PMID:36136483 ![]()
RAB6 and dynein drive post-Golgi apical transport to prevent neuronal progenitor delamination
Brault JB, Bardin S, Lampic M, Carpentieri JA, Coquand L, Penisson M, Lachuer H, Victoria GS, Baloul S, El Marjou F, Boncompain G, Miserey-Lenkei S, Belvindrah R, Fraisier V, Francis F, Perez F, Goud B, Baffet AD
EMBO Rep. 2022 Aug 18:e54605
PMID:35979738 ![]()
Novel role of the synaptic scaffold protein Dlgap4 in ventricular surface integrity and neuronal migration during cortical development
Romero DM, Poirier K, Belvindrah R, Moutkine I, Houllier A, LeMoing AG, Petit F, Boland A, Collins SC, Soiza-Reilly M, Yalcin B, Chelly J, Deleuze JF, Bahi-Buisson N, Francis F
Nat Commun. (2022) 13, 2746
PMID:35585091 ![]()
Visualising the cytoskeletal machinery in neuronal growth cones using cryo-electron tomography
Atherton J, Stouffer M, Francis F, Moores CA
J Cell Sci. (2022) 135
PMID:35383828 ![]()
Doublecortin mutation leads to persistent defects in the Golgi apparatus and mitochondria in adult hippocampal pyramidal cells
Stouffer MA, Khalaf-Nazzal R, Cifuentes-Diaz C, Albertini G, Bandet E, Grannec G, Lavilla V, Deleuze JF, Olaso R, Nosten-Bertrand M, Francis F
Neurobiol Dis. (2022) 168, 105702
PMID:35339680 ![]()